




What are FDA IND applications? An IND, or Investigational New Drug, application demonstrates to the Food & Drugs Administration that your medicinal product is safe, compliant and suitable for testing in humans. A successful IND application means your investigational product is approved for use in clinical trials and for transportation across state lines within the United States. The IND is a crucial milestone for a pharmaceutical company that seeks to bring a new drug into the American market.
For support on your way to clinical and market approval, you may find it useful to get guidance via a pre IND meeting with the FDA. They can support you at the critical ‘Pre-Clinical Development’ stage of the application.
What is a pre IND meeting FDA?
Pharmaceutical companies that are going through the IND application process are allowed to have meetings with the FDA to discuss the development and review of their applications. The meeting is known as the 'pre IND meeting' with the FDA. This is an opportunity for the applicant to discuss any scientific, regulatory or safety issues related to the development of a new drug.
The pre IND meeting allows the applicant or sponsor to ask specific questions concerning
- their initial US clinical study (known as the first-in-human (FIH) study for new drugs and biologics)
- the nonclinical program
- manufacturing
- product quality
- regulations
- anything else that is relevant to the IND application

Types of pre-IND FDA meeting
The FDA offers two programs to guide and accelerate your IND processes:
- the INitial Targeted Engagement for Regulatory Advice on CBER producTs (INTERACT) Program
- the Pre-Investigational New Drug Application (IND) Consultation Program
These programmes are designed to establish an effective working relationship between your organisation and the FDA, and will usually make the IND submission and review stages quicker and easier.
Key benefits of a pre IND meeting with the FDA
- The pre IND meeting with the FDA is a free service
- The pre IND meeting can help improve an applicant's speed to market because the FDA can help them to complete the IND application properly, avoid common mistakes, and mitigate the possibility of clinical holds.
- It allows applicants to to gain recommendations from the FDA and form a preliminary agreement on elements of the drug development program.
- Build rapport with key contacts at the FDA.
FDA pre IND meeting guidance: how to request a meeting
First, applicants will need to ensure they have the details of the FDA review division that is responsible for reviewing the IND application. The review divisions are organized generally along therapeutic class. If you want to check which division you need, there are contact numbers for the different divisions available here.
The following information will be needed:
- Product name, application number (if applicable), chemical name and structure
- Proposed indication(s) or context of product development
- Type of meeting required (i.e., Type A, Type B, or Type C). Include a rationalle for the type of meeting chosen.
- A brief statement which details the purpose and objectives of the meeting
- A proposed agenda
- A list of proposed questions grouped by discipline. Alongside each question, provide a supporting statement which outlines its context and purpose.
- Names of all individuals who will attend the meeting, including any consultants. Include their titles and affiliations.
- Names of any FDA staff, if appropriate, who should participate in the meeting.
- Any suggestions for dates and times for the meeting.
- Details concerning the format of the meeting (e.g, in person, online etc.).
Stating the purpose and objectives of the pre IND meeting FDA
Sponsors and applicants should ensure that this statement is clear and succinct. For example, a small table could be included to summarize key results. Do not provide lengthy documentation concerning trial designs or clinical/ nonclinical studies.
The statement should include:
- Details of any completed or planned studies (clinical and nonclinical)
- A brief summary of the context and issues
- Relevant data or clinical trials to be discussed
- General overview of critical questions to be explored at the meeting
- How the pre IND meeting fits in to wider development plans
The FDA's response and timelines for pre IND meetings with the FDA
After the request for a meeting has been filed, the FDA will decide whether or not to arrange a pre IND meeting. They will also decide on the format of the meeting. The FDA will typically respond within 21 days and meetings usually take place within 60 days of the request. Based on this timeline, applicants should submit their pre IND requests about two months in advance of their desired meeting date.
Types of FDA IND application
There are multiple types of IND application. Broadly speaking, your IND will be either 1) commercial or 2) research. It depends on whether you intend to submit a marketing application afterwards or not.
There are then three IND ‘types’ underneath this broad categorisation:
- Investigator IND, for investigation of an unapproved drug. This is the most common IND type.
- Emergency use IND, for quick authorisation of a drug for use in an emergency situation (such as the COVID-19 pandemic) when no alternative treatment exists
- Treatment IND, for drugs already in the clinical stage which are already proving effetive in treating serious conditions
Around a third of IND applications relate to biopharmaceutical products. Two thirds relate to small-molecule drugs.
Filing an IND: when should you begin your IND process?
Ultimately, the IND stage begins once your pre-clinical activities are concluded. That means you should:
- Be confident in the strength and integrity of your product performance, and ready to take it into the human testing stage.
- Have sufficient proof-of-concept data to present to the FDA to attest to your product’s safety and efficacy so far.
The IND submission process is like any other regulatory hurdle. You’ll need documented information, and lots of it. Replicable results, performance hypotheses and activity analytics are all solid and reassuring data that the FDA will expect to see in tandem with demonstrable GxP operation on your part.
Key pieces of IND submission data include:
- Toxicology and pharmacology data from animal testing
- Storage and stability data
- Manufacturing information showing how you intend to manufacture your product at scale and with sufficient quality
- Clinical research plans and clinical protocols to assess potential risk to humans
- Target clinical indication to contextualise your submission
GMP (Good Manufacturing Practice) and GLP (Good Laboratory Practice) benchmarks should underpin all the data you submit to the FDA.
IND submissions
So you have all your pre-clinical data and are ready to proceed to clinical development? The IND submission stage begins.
Your collected data should be submitted in triplicate to the FDA alongside three supplementary forms.
The FDA then has 30 days to review and approve your product. In the case of approval, or simply a lack of rejection in the 30-day timeframe, your product has the green light to progress to the clinical testing stage and may be transported to clinical investigators in different states.
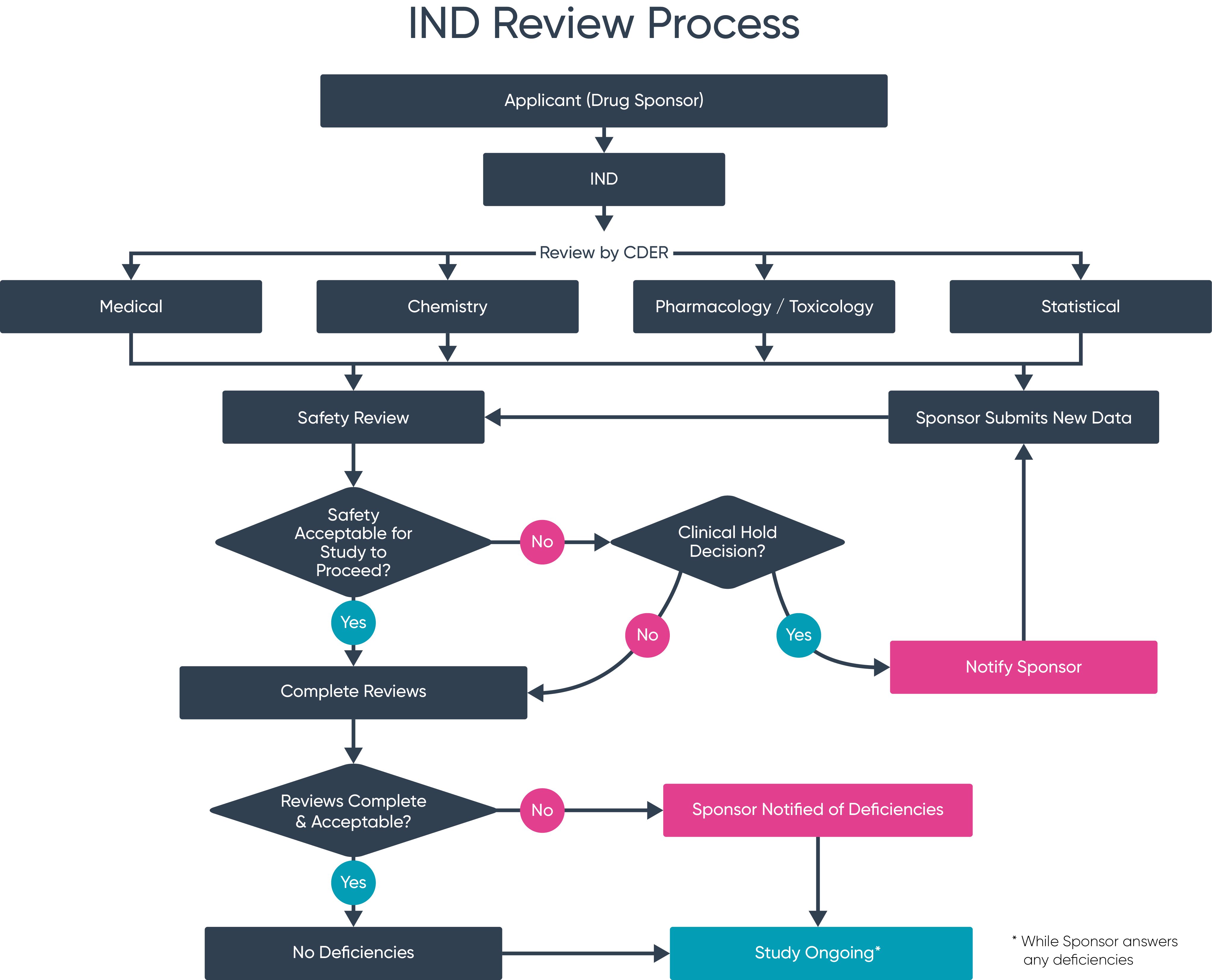
Managing your IND application
Managing, controlling and co-authoring your IND data pack can be a long and complex task that distracts you from your core product development work.
Needless to say, organisations going through the IND submission process do make mistakes. Common errors have emerged as recurring blockers of the process.
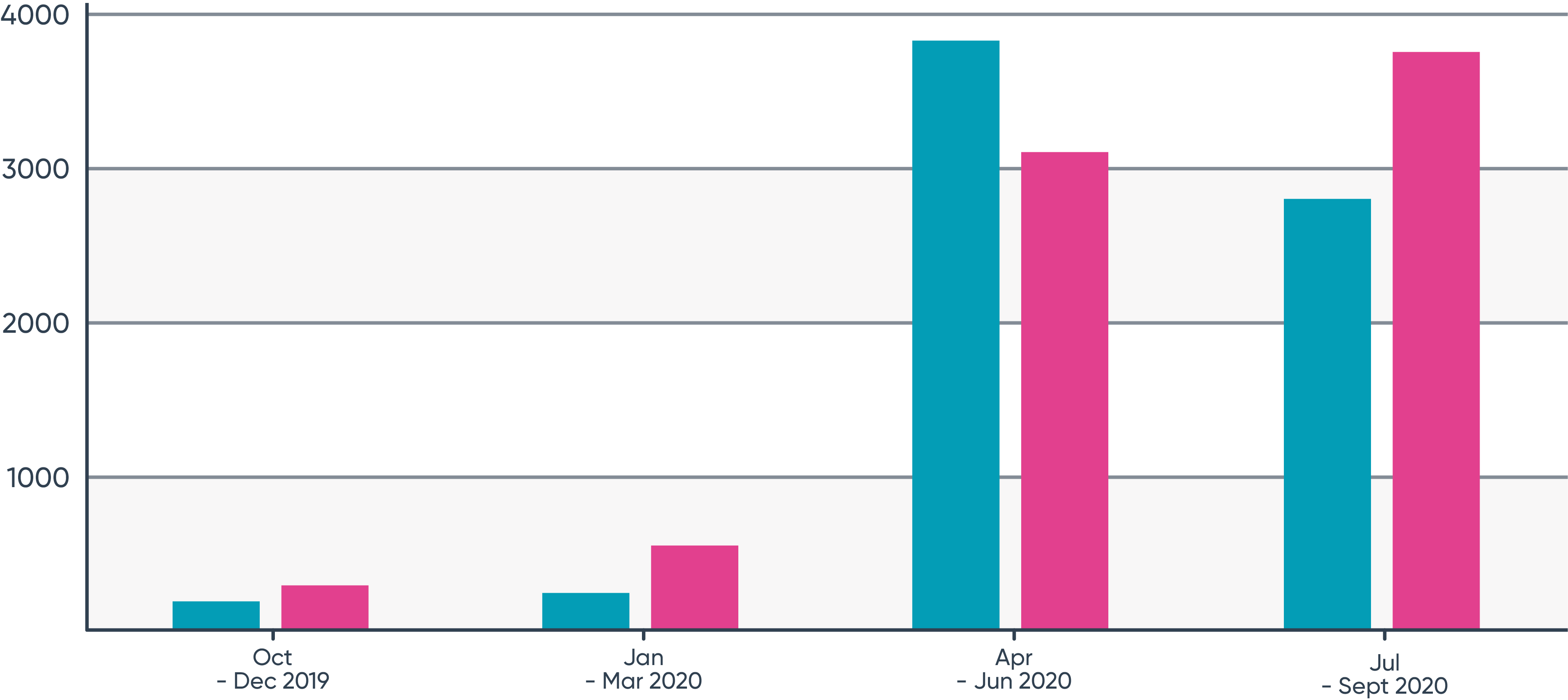
As we can see from this graph, in the FDA’s 2020 fiscal year it received 6,954 IND submissions (blue) and took 7,603 actions (yellow) against them – meaning that on average, every single IND submission required a corrective FDA response within the 30-day timeframe, with about 9% requiring more than one response.
A controlled, right-first-time approach is therefore a great way to impress the FDA and accelerate your route to the clinical stage.
In the past five years, the three most common reasons for IND data pack rejections have been:
- Submission mistakes: missing documents or incorrect eCTD formats
- Disorganised and unclear data that the FDA simply cannot process and digest within 30 days
- Unfocused and irrelevant data that doesn’t support your clinical protocol or adequately explain potential risks
These mistakes are compounded and multiplied by outdated document creation and review techniques, with multiple uncontrolled versions circulating simultaneously by email or spreadsheet. For multi-site and international organisations, coordinating relevant stakeholders and establishing a clear and cogent route to submission can be a serious challenge.
Reduce your IND document review cycle by up to 65% with PleaseReview
Document collaboration systems such as PleaseReview are becoming increasingly adopted and invaluable tools for life science organisations looking for effective and painless IND submissions.
Key benefits of PleaseReview
- Collaborate on single, centralised, shared versions of IND documents, wherever you’re based.
- Flag mistakes, fix omissions and have in-text conversations in real-time.
- Access complete audit trails and histories of all document changes and revisions.
- Proven to reduce document review cycles by on average 65% (on average)
We hope you have found our guide to pre IND meeting FDA useful. Discover Ideagen's expertise in helping pharmaceutical companies to improve their speed to market with our document collaboration solution for IND applications.
Accelerate your FDA IND application with PleaseReview
Join the industry standard and discover how PleaseReview could transform the way that your company collaborates on complex documents such as FDA IND applications.