




If you’re a medical device company operating in the US market, 21 CFR Part 820 is a Food and Drug Administration (FDA) regulation you’ll need to be familiar and compliant with. CFR stands for the Code of Federal Regulations and title 21, known as 21 CFR, is a set of rules for all things food and drug related. We’ve put this guide together to break down the different parts of this key regulation, outlining what you need to know and why compliance is important.
What is 21 CFR Part 820 and why should you care?
21 CFR Part 820 is part of the Current Good Manufacturing Practice (CGMP) regulations. It ensures that all medical devices created and developed within the US market are safe and follow satisfactory quality processes at all stages of development. This is important for manufacturers to ensure they are producing medical devices which are effective and assure patient safety.
This regulation is a requirement for all medical device manufacturers. Being subject to inspection from the FDA, non-compliance or being found to fall short of the minimum standards will result in receiving a warning letter which will also be publicly shared on the FDA website. This means that distributers and patients can access this information to alert them of any possible safety concerns with their medical devices. As a manufacturer, this is of course something that you want to avoid as the reputational damage has the potential to be long lasting and negatively impact your performance within the market.
What is the difference between ISO 13485 and 21 CFR 820?
ISO 13485 is the international standard for a quality management system for medical devices. It’s not mandatory, but meeting it helps to maximise quality and to showcase manufacturers dedication to producing high quality devices which prioritise patient safety. 21 CFR 820 is specific to medical device manufacturers operating in the US, or the US market. While it also specifies what’s required from a quality management system tailored to the development of medical devices, 21 CFR 820 is a requirement mandated by the FDA.
While there are similarities between the two, the main differences are:
- 21 CFR 820 is mandatory, whereas ISO 13485 is voluntary
- 21 CFR 820 only applies to the US
- ISO 13485 follows a risk-based approach
- ISO 13485 follows an updated structure, whereas 21 CFR 820 has been laid out in the same way since 1997
- 21 CFR 820 is the product of the FDA, whereas ISO 13485 was created collaboratively
It is planned for the FDA to adopt ISO 13485 as their regulatory framework in place of the older 21 CFR 820. The mapping out of this transition is still underway, so no official dates or timelines have been confirmed by the FDA yet. This means in the future, manufacturers will use ISO 13485 as their regulatory framework but until this change takes place, 21 CFR 820 remains in place.
FDA 21 CFR 820: what it includes and how to comply
FDA 21 part CFR 820 is made up of 15 subparts- each one focusing on a particular area within medical device manufacturing. Below we summarise each subpoint and explain what you need to do to make sure you are compliant.
A- General provisions
This part of the regulation is all about providing definitions and outlining who it’s applicable to in line with the Federal Food, Drug, and Cosmetic Act. The main takeaway from this part is that 21 CFR part 820 is applicable to any medical device either manufactured in the US or imported in and intended for use in the US.
B- Quality system requirements
In this section, the FDA outline all their requirements for a quality management system and what you should consider when implementing one. Executive level managers are expected to put in place the necessary policies and ensure this is communicated and understood throughout the organisation. This includes establishing who is responsible for what and structuring accordingly, alongside managing training and employee performance.
The quality management system should:
- Follow established and documented procedures
- Contain a quality plan for each part of the manufacturing process
- Enable management to review quality activities and report on its performance
C- Design controls
This specifies the need to have controls in place for each section of the design process. This takes into account each step from planning all the way through to validation and review. This is applicable to all class II or III devices, as well as many class I devices.
Your quality management system should cover the following steps:
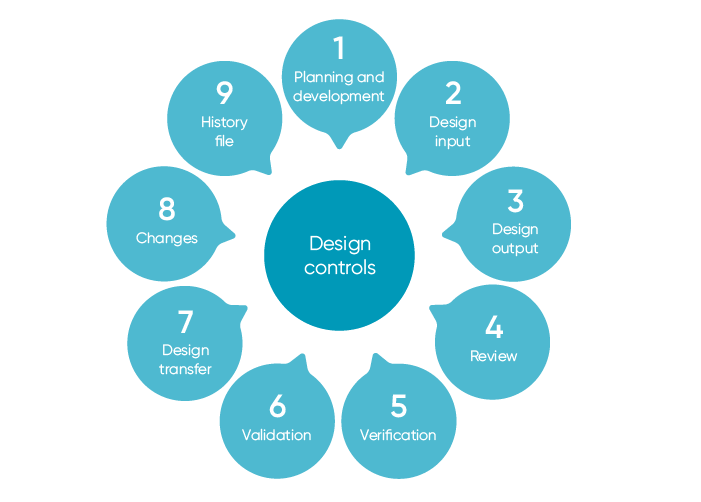
D- Document controls
Document management is a key aspect of your quality process. You’ll need to have a system in place which can enable approval and distribution of key documents, ensuring they are both secure and accessible when needed. There should also be a process for changes and updates to be communicated and circulated.
E- Purchasing controls
This is all about managing your supply chain and any contractors and consultants you work alongside. You’ll need to review potential suppliers or contractors to ensure they meet the right quality standards, put in place a process for working with them, keep records and manage purchasing data.
F- Identification and traceability
Any finished device must clearly show who made it, and also state the make and model. This ensures that should any fault occur, the patient or healthcare provider can report this and the right action can be taken.
G- Production and process controls
This is one of the meatier subparts which outlines everything that needs to be thought about to make sure the finished medical device contains all the required specifications. This also includes putting in place appropriate corrective actions to ensure this happens.
The key things to think about here include:
- Documenting instructions and SOPs (standard operating procedures)
- Having a control in place for each step of the production process
- Ensuring all standards and regulations are complied with
- Validate and ensure the safety of all equipment and processes
- Having adequate monitoring and inspection activities throughout
The full standard breaks down every aspect that needs to be considered within these controls, such as the surrounding environment, hygiene and handling controls, the use of equipment and safety of the materials used.
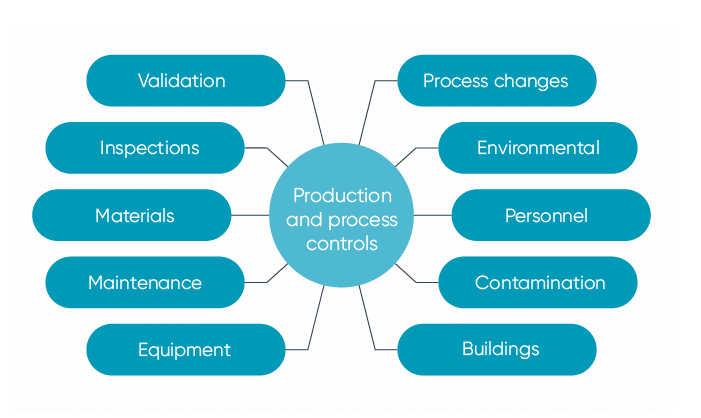
H- Acceptance activities
These are your inspections and validation processes- so your quality management system should incorporate how you conduct testing, inspections, and verification activities.
I- Non-conforming product
While you’ll obviously be taking steps to ensure that the devices you manufacture are up to standard, you’ll need to have procedures in place in case a finished product doesn’t conform to the correct standards. There are several steps you’ll need to address to ensure all necessary elements are considered:
- How to identify a non-conforming device within the inspection stage
- A procedure for documenting it
- Evaluating what went wrong
- Procedure for additional work
J- Corrective and preventative action
This is your CA/PA processes where you map out corrective and preventative actions for all possible risks and errors. These should be created for all areas including processes, audits, records and dealing with complaints and returns.
Each CA/PA process should look like this:
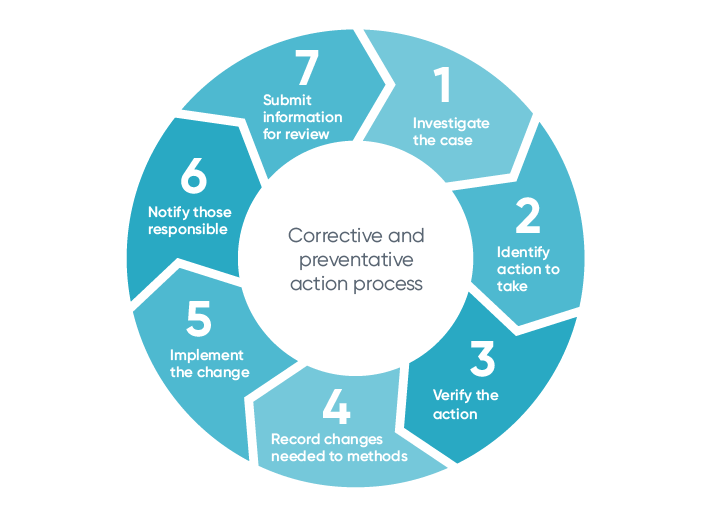
K- Labelling and packaging control
These are controls to ensure that your labels are clear and affixed properly to the device so that healthcare professionals are able to clearly identify the information they need. You’ll need to plan out a set of procedures for producing and inspecting labels to ensure they contain the correct information and are placed on the right product.
L- Handling, storage, distribution and installation
This is about ensuring that mistakes don’t happen while the finished products are being handled and moved. Consider it this way- you’ve put all this time and effort into all the other controls and procedures so far, so if an undesirable event occurred now, it would negate all that hard work. Quality extends beyond the manufacturing of your device. It’s about assuring the safety of them at all stages and making sure the individuals installing or using the devices have clear guidelines on how to do this.
M- Records
There are specific records that need to be kept and made available for FDA inspection. These include:
- Device master record- this should contain all necessary information such as the device and production specifications; quality assurance procedures; labelling and packaging details, and installation, maintenance, and servicing instructions.
- Device history record- this will include key dates, quantities manufactured and distributed and identification codes
- Quality system record- this should detail the location of where procedures and activities are stored
- Complaint files- all details should be recorded, including the investigation and outcome of a complaint
N- Servicing
If servicing is a requirement for the medical device/s you are producing, there should be detailed instructions for both validating and carrying this out. Records also need to be kept detailing when servicing takes place and who carried it out- including the specifics of what was done.
O- Statistical techniques
If needed, statistical techniques should be put in place to establish, control and verify the capability of your processes and the product characteristics. This is to analyse your data and ensure your controls are effective.
The benefits of adopting a good quality management system
While there’s a lot to being compliant with 21 CFR part 820, all of these components can be incorporated into your quality management system. This ensures you are meeting the FDA requirements but beyond that it allows you better manage everything so that you can consistently produce high quality products with less chance of mistakes occurring.
The right quality management system can:
- Help you to comply with the FDA requirements
- Provide a central place to manage all the necessary processes, records and documentation
- Allow you to easily access key information when needed
- Improve the quality of your manufacturing process and finished products, helping to establish trust within the market
- Prevent costly mistakes
How software can help comply with 21 CFR part 820
It’s a lot of work to ensure that you are meeting every part of the FDA regulation. Using dedicated software gives you the central system that’s needed in order to manage and gain an overview of all the different areas and processes that are in place. Documents, audits and reports are all digital and easily accessed and it allows you to better manage workflows and required actions.
Explore quality management solutions
Automate and streamline your quality processes, identify opportunities for excellence and achieve compliance with regulations and standards.