
CTIS receives record number of clinical trial applications
Since the new Clinical Trial Information System (CTIS) was introduced on 31 January 2022, the number of clinical trial applications (CTAs) has steadily grown over six months. While nine applications were received in the first month, July saw a record number of 58 submissions, which is a marked increase on June’s total of 39.
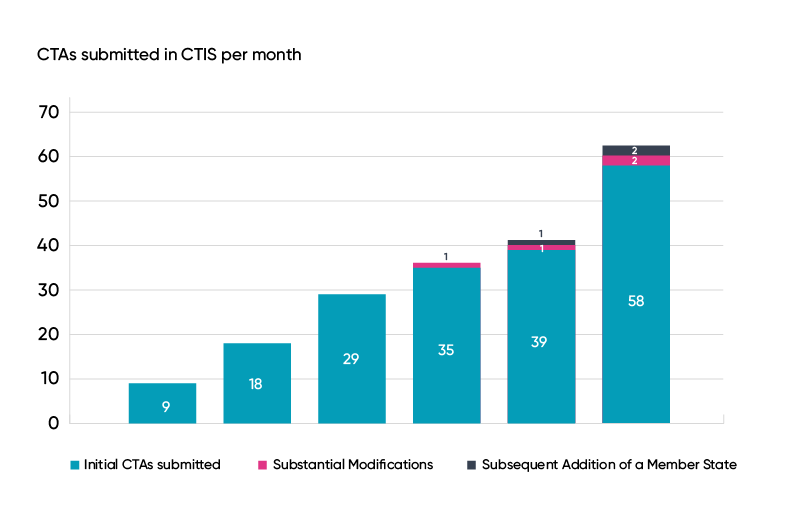
Data sourced from ‘Key performance indicators (KPIs) to monitor the European clinical trials environment’ report, EMA, 23rd August 2022.
According to the European Medicine’s Agency (EMA), nearly 195 CTAs have been submitted since the launch of the new system since 31 January. However, this represents less than half of all applications overall. Over the six-month period, 224 applications were registered using the old database: the EudraCT. Since the launch of the CTIS, the three countries with the highest number of applications to the new system are Denmark, Spain and Belgium.
When will the CTIS replace EudraCT?
While use of the CTIS is not yet mandatory for clinical trial sponsors, the system will officially replace the EudraCT portal in January 2023. Sponsors will be obliged to use the CTIS to apply for authorisation to run a clinical trial.
Of the 195 CTAs submitted to the CTIS, 188 are initial clinical trial applications, four are substantial modification applications and three include substantial additions from concerned Member States.
What is the EU CTIS for?
In simple terms, the CTIS was developed so that sponsors could use apply to run simultaneous clinical trials throughout the European Union using the same documentation. Clinical trial sponsors can apply for clinical trial authorisation for up to 30 European countries with one application
CTIS serves to implement EU pharmaceutical law in the Clinical Trials Regulation (Regulation (EU) No 536/2014).The European Medicines Agency (EMA) EMA maintains CTIS and the public website, together with the EU Member States, EEA countries and European Commission.
CT Regulation will fully replace the former Clinical Trials Directive (2001/20/EC) (“CT Directive”) and the implementing laws in the member states.
Who does it apply to?
The CTR will apply to all clinical trials that are conducted within the European Union.
The CTR will not apply in the UK and it remains to be seen whether the UK will elect to align its legislation with the CTR. In any event, the UK will not be able to benefit from the advantages of being part of the CTIS.
What are the benefits of using the CTIS?
According to EMA, sponsors should submit their applications to the new portal for four key benefits:
1. Improved information sharing
2. Collective decision-making on clinical trials
3. Increased transparency on clinical trials
4. Ensuring high safety standards for patients
Decisions made on CTIS applications since January
- 43 decisions have been made so far on CTAS from both commercial sponsors and non-commercial entities.
- 12 decisions related to applications to market the treatment in one member state.
- Nine decisions were rendered on applications to market a drug in multiple nations.
- 16 were for Phase II trials.
- 10 were for Phase IV trials.
- Six were for Phase III trials.
What are the main therapeutic areas so far?
The following areas have had decisions issued by the CTIS since 31 January:
- Neoplasm treatments
- Treatments for viruses
- Treatments for cardiovascular diseases
- Diseases of the skin and connective tissues
- Nutritional and metabolic diseases
- Musculoskeletal diseases
- Immune system diseases
- Digestive system diseases
Transparency and disclosure requirements for CTIS applications
According to the draft guidance from the EMA, medical writers ‘can play an important role in reducing the need for redactions’ in documentation submitted to the CTIS. They request that sponsors make documents as transparent and accessible as possible, even if this means publication is deferred.
Guidance from the EMA states that ‘[i]t is expected that embedding a CCI identification and tracing strategy during the writing of the CTIS related documentation would limit the unnecessary dissemination of commercially confidential information in documents where these pieces of information are not essential, required or relevant.’
-
Identifying CCI early on and minimising its distribution throughout the document
The EMA suggests that ‘the sponsors and marketing authorisation applicants/holders consider identifying early during the development plan those pieces of information which are considered CCI, track these as the product evolves and proactively minimise the distribution of these pieces of information across the clinical trial documentation already when the documents are written.’
-
Using document templates
The tracing strategy can be further complemented by employing document templates which specifically indicate which information is required to be included in the documents according to the clinical trial legislation, scientific guidelines and regulatory guidance.
-
Tagging
As a complementary approach, tagging those pieces of information which are considered CCI at the time the clinical trial documents are written would facilitate the preparation of the document versions meant to be published.
It is envisaged that implementing such approaches would reduce the efforts entailed by preparing separate document versions for publication purposes and it would allow the CTIS users to publish in higher proportion the very same documents which were submitted for scientific evaluation.
How technology can help
While preparing documents for the CTIS with transparency and disclosure requirements in mind is a time-consuming and costly process, Ideagen’s PleaseReview software can help streamline the entire process from start to finish.
Ideagen PleaseReview is a document co-authoring, review and redaction platform used widely by medical writing teams, regulatory affairs departments, clinical research organisations and more.
Key benefits of PleaseReview for clinical trials sponsors using the CTIS:
- High volume teams can work in real-time on complex document reviews in a stable and robust review environment.
- Work in real-time on PDF documents.
- Its advanced redaction capabilities make it easy to comply with CTIS rules and guidance concerning the transparency and disclosure of data published for the public.
- The real-time collaborative environment also helps teams to identify CCI from the start and work together to reduce the need for anonymisations and redactions across the document.
- Easily tag, and categorise, data.
- An automatic reconciliation report captures all document activity, which makes it easy to see what was redacted or anonymised—when, where and by whom.
- Eases the administrative burden on those responsible for preparing the document for publication to the public
- Quickly call out someone – query use of PPI/ CCI. Make suggestions for how the data can be anonymised
- Integrates with Veeva Vault
- Set up review templates to speed up review creation

Success stories
Read how UniQure created a structured and consistent process for document development with PleaseReview.